L’amyotrophie spinale infantile
L’amyotrophie spinale en quelques mots
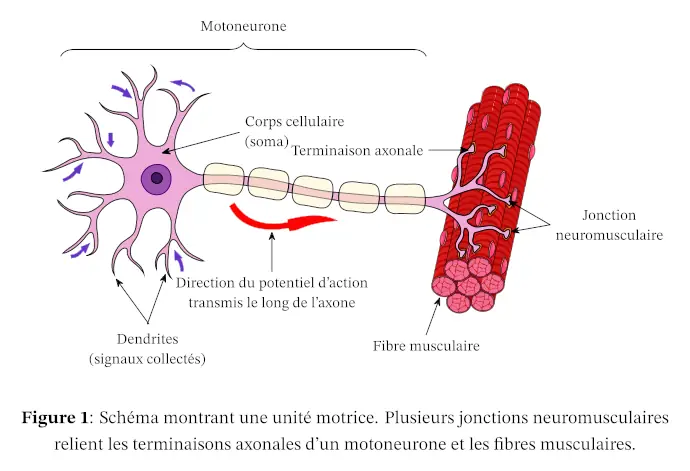
Les amyotrophie spinales infantiles, car il en existe plusieurs formes, sont dues à la dégénérescence de certaines cellules nerveuses de la moelle épinière appelées motoneurones.
Ces motoneurones permettent la transmission des informations entre le cerveau et les muscles .
En l’absence de ces cellules, les nerfs moteurs des muscles sont lésés et meurent, ils n’acheminent plus l’ordre du mouvement jusqu’aux muscles.
Les conséquences de l’amyotrophie spinale
Les muscles concernés sont ceux du bassin, des épaules, du tronc et des membres (inférieurs et supérieurs). Devenus inactifs, les muscles s’affaiblissent, s’atrophient et se rétractent.
Selon l’âge d’apparition, les premières difficultés motrices pourront se traduire par l’absence d’acquisition de la station assise, peu de gesticulation spontanée, l’absence d’acquisition de la marche, des difficultés à tenir la tête …
Dans les formes les plus graves, les muscles intercostaux sont paralysés pouvant engendrer une assistance ventilatoire à un jeune âge.
L’amyotrophie spinale étant de mieux en mieux connue, 3 traitements sont désormais sur le marché. Aucun ne permet encore de guérir, mais ils permettent un ralentissement de l’évolution de la maladie et l’un d’eux, prit dès le diagnostique permet un développement moteur impressionnant.
Pour plus d’infos

Soutenez l’association
Vous pouvez faire la différence et soutenir Alyssa ! En parlant de la maladie autour de vous, en participant aux différents événements organisés par l’association ALYSSA 63, en adhérant à l’association ou en faisant un don, que vous soyez un particulier ou une entreprise.